Using MPD's SNP retrieval utility
Chromosomal coordinates are GRCm38 / mm10. Functional annotation is from dbSNP build 142.
If you wish, you can select a specific data set from the pulldown.
Or, you can let it be determined automatically based on strains you select.
We're prepared to load
additional SNP data sets based on community interest.
Any call cells that are white / empty indicate "no data available".
Click on Include additional flank to add upstream and/or downstream flank to the retrieval basepair range for each requested gene or marker.
When searching on gene / marker symbols, MGI's current nomenclature is recognized, and retrieved locations are based on MGI's current coordinates. MGI batch query can be used to get current gene symbols. Please note that this MGI gene information sometimes differs from the dbSNP 142 SNP annotation.
You can also have two or more strains in Group A and/or Group B, in which case a given location is considered polymorphic if all useable calls in Group A differ from all usable calls in Group B (the calls in Group A must be uniform and likewise for Group B). Cells that are "No data" or "Het" are ignored; if a group ends up with no usable calls then that location is ineligible for comparison.
abbreviated notation:
code : gene
Certain function codes (Cn, Cs) have additional information appended to this construct, see below for more info.
The dbSNP 142 annotation gene names/locations sometimes differ from the MGI gene information found elsewhere in MPD.
Some basepair locations have multiple, differing annotations due to more than one transcript covering the location. In these cases MPD lists the above construct for each unique instance seen, separated by a space. On the other hand, some basepair locations have no functional annotation at all, either because the location is not within any gene, or because the location is not present in NCBI dbSNP mouse build 142. You can click on the dbSNP RS number linkout for further details on these annotations.
Basic usage
On MPD's SNP data retrieval utility page, specify genomic region, desired mouse strains or data set, and any desired filtering, then . You'll be able to preview your result on the web page and download your entire result as CSV. If you'd like to adjust and rerun your query just use your browser's Back button to return to the form.Chromosomal coordinates are GRCm38 / mm10. Functional annotation is from dbSNP build 142.
Available data sets
| Data set | Procedure | What's in this data set | Panel | Sex | Age (weeks) | Year |
|---|---|---|---|---|---|---|
|
|
genotyping | SNP profiling, 131,000+ genomic locations, 1-19,X. | inbred (89) | 2009 | ||
|
|
genotyping | SNP profiling, 470,000+ genomic locations, 1-19,X,Y,MT. | inbred (142) | m | 2014 | |
|
|
genotyping | SNP profiling, 470,000+ genomic locations, 1-19,X,Y,MT. | BXD w/par (92) | m | 2014 | |
|
|
genotyping | SNP profiling, 470,000+ genomic locations, 1-19,X,Y,MT. | ILSXISS w/par (69) | m | 2014 | |
|
|
genotyping | SNP profiling, 470,000+ genomic locations, 1-19,X,Y,MT. | AXB, BXA, BXH, CXB, AKXL w/par (72) | m | 2014 | |
|
|
genotyping | SNP profiling, 470,000+ genomic locations, 1-19,X,Y,MT. | B6.A, B6.PWD consomic panels (53) | m | 2014 | |
|
|
genotyping | SNP profiling, 8,100,000+ genomic locations, 1-19,X,Y,MT. | inbred (16) | m | 2005 | |
|
|
genotyping | SNP profiling, 80,000,000+ genomic locations. SNPs and indels. 1-19,X. | inbred (37) | both | 2017 | |
|
|
genotype assessment | 70+M SNPs, 7.5+M indels, 573k structural variants | inbred (39) | 2025 | ||
|
|
genotyping | SNP profiling, 132,000+ genomic locations, 1-19,X | HMDP (248) | both | 2018 | |
|
|
genotyping | SNP profiling, 130,000+ genomic locations, 1-19,X,Y,MT. | CC w/par (77) | m | 2020 | |
|
|
genotyping | SNP profiling, 76,000+ genomic locations, 1-19,X,MT | CC w/par (77) | both | 2017 |
Please contact us to register interest in any of the following data sets:
Chicago1 8200+ locations, 58 inbred strains (2009)
CNB1 1300+ locations, 18 inbred strains. (2010)
JAXSNP1 2000+ locations, 107 inbred strains (2007)
UNC-MUGA1 7400+ locations, 19 inbred strains (2011)
WUSTL1 2300+ locations, 16 LGXSM strains (2006)
Chicago1 8200+ locations, 58 inbred strains (2009)
CNB1 1300+ locations, 18 inbred strains. (2010)
JAXSNP1 2000+ locations, 107 inbred strains (2007)
UNC-MUGA1 7400+ locations, 19 inbred strains (2011)
WUSTL1 2300+ locations, 16 LGXSM strains (2006)
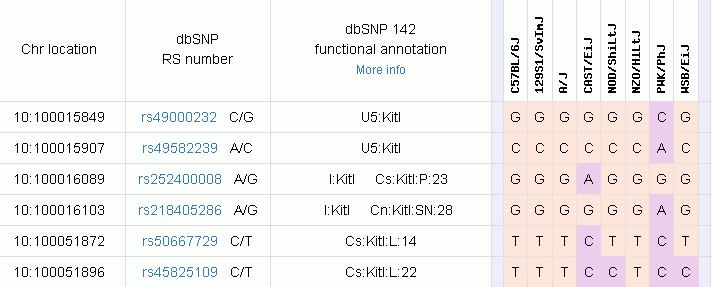
Screen shot of example result
Any call cells that are white / empty indicate "no data available".
Specifying genes | markers | regions
Enter a gene symbol or chromosome coordinate range. Click on the Show examples link to see various possibilities. As noted there, multiple items can be supplied, and it's also possible to retrieve an entire chromosome or entire genome (except for the largest data sets).Click on Include additional flank to add upstream and/or downstream flank to the retrieval basepair range for each requested gene or marker.
When searching on gene / marker symbols, MGI's current nomenclature is recognized, and retrieved locations are based on MGI's current coordinates. MGI batch query can be used to get current gene symbols. Please note that this MGI gene information sometimes differs from the dbSNP 142 SNP annotation.
Here's how to use MGI batch query to be sure all symbols in your set of genes are current:
1. Go to MGI batch query
2. Copy-paste your gene list into the tool
3. For Type, select "All Symbols/ Synonyms/ Homologs"
4. Check the box for Genome Location
5. Click Search
6. On the result page click on Excel File
7. Open the Excel result and see column D
8. Copy-paste column D to your destination.
1. Go to MGI batch query
2. Copy-paste your gene list into the tool
3. For Type, select "All Symbols/ Synonyms/ Homologs"
4. Check the box for Genome Location
5. Click Search
6. On the result page click on Excel File
7. Open the Excel result and see column D
8. Copy-paste column D to your destination.
Choosing mouse strains
If you selected a specific data set you'll get a pulldown of available strains. Otherwise you'll be typing into an input field with search-suggest. Either way, after choosing a strain click on the green button to add it to the query. If you're not sure about strains, type inCC8 which provides a good default.
Polymorphism filtering with strain groups A and B
The most common use of this feature is to limit your result to have only the locations where the call for one strain differs from that of another. To do this choose a strain for Group A and a different strain for Group B. If either strain's call is "No data" or "Het" then that location is ineligible for comparison.You can also have two or more strains in Group A and/or Group B, in which case a given location is considered polymorphic if all useable calls in Group A differ from all usable calls in Group B (the calls in Group A must be uniform and likewise for Group B). Cells that are "No data" or "Het" are ignored; if a group ends up with no usable calls then that location is ineligible for comparison.
Interpreting dbSNP 142 functional annotation
Using a variation effect prediction algorithm, NCBI dbSNP has annotated basepair locations within genes as intronic, coding, or several other classes. In your results, MPD represents these annotations using anCertain function codes (Cn, Cs) have additional information appended to this construct, see below for more info.
The dbSNP 142 annotation gene names/locations sometimes differ from the MGI gene information found elsewhere in MPD.
Some basepair locations have multiple, differing annotations due to more than one transcript covering the location. In these cases MPD lists the above construct for each unique instance seen, separated by a space. On the other hand, some basepair locations have no functional annotation at all, either because the location is not within any gene, or because the location is not present in NCBI dbSNP mouse build 142. You can click on the dbSNP RS number linkout for further details on these annotations.
| MPD function code | dbSNP equivalent term |
Definition and Sequence Ontology link |
| Cn | missense | "Coding nonsynonymous". A sequence variant, that changes one or more bases, resulting in a different amino acid sequence but where the length is preserved. SO:0001583 In MPD result displays, two amino acid codes (reference and variant) are appended to this annotation, followed by the amino acid position. |
| Cs | synonymous-codon | "Coding synonymous". A sequence variant where there is no resulting change to the encoded amino acid. SO:0001819 In MPD result displays, one amino acid code (the reference) is appended to this annotation, followed by the amino acid position. |
| U5 | utr-variant-5-prime | A UTR variant of the 5' UTR. SO:0001623 |
| U3 | utr-variant-3-prime | A UTR variant of the 3' UTR. SO:0001624 |
| I | intron-variant | A transcript variant occurring within an intron. SO:0001627 |
| NC | nc-transcript-variant | A transcript variant of a non coding RNA gene. SO:0001619 |
| StopG | stop-gained | A sequence variant whereby at least one base of a codon is changed, resulting in a premature stop codon, leading to a shortened transcript. SO:0001587 Amino acid info is shown similarly to Cn above. |
| StopL | stop-lost | A sequence variant where at least one base of the terminator codon (stop) is changed, resulting in an elongated transcript. SO:0001578 Amino acid info is shown similarly to Cn above. |
| SSA | splice-acceptor-variant | A splice variant that changes the 2 base region at the 3' end of an intron. SO:0001574 |
| SSD | splice-donor-variant | A splice variant that changes the 2 base region at the 5' end of an intron. SO:0001575 |
| Cf | frameshift-variant | (indels only) An attribute describing a sequence that contains a mutation involving the deletion or insertion of one or more bases, where this number is not divisible by 3. SO:0000865 |
| Xi | cds-indel | (indels only) An indel variation with length of multiple of 3bp, not causing frameshift (no SO term) |
Including indels in the result
One data set (Sanger) includes indels (small insertions or deletions that are larger than the usual one basepair size of SNPs). You can opt to include these in your result using the checkbox near the bottom of the form. MPD does not retain specific allele sequences for indels. Rather we encode them so that the C57BL/6J reference is always0
then the other reported variants are assigned 1, 2 and so on. To see actual allele sequence use the
linkout to Sanger VCF.